| 红白血病 | 您所在的位置:网站首页 › find af randomised研究提示 › 红白血病 |
红白血病
|
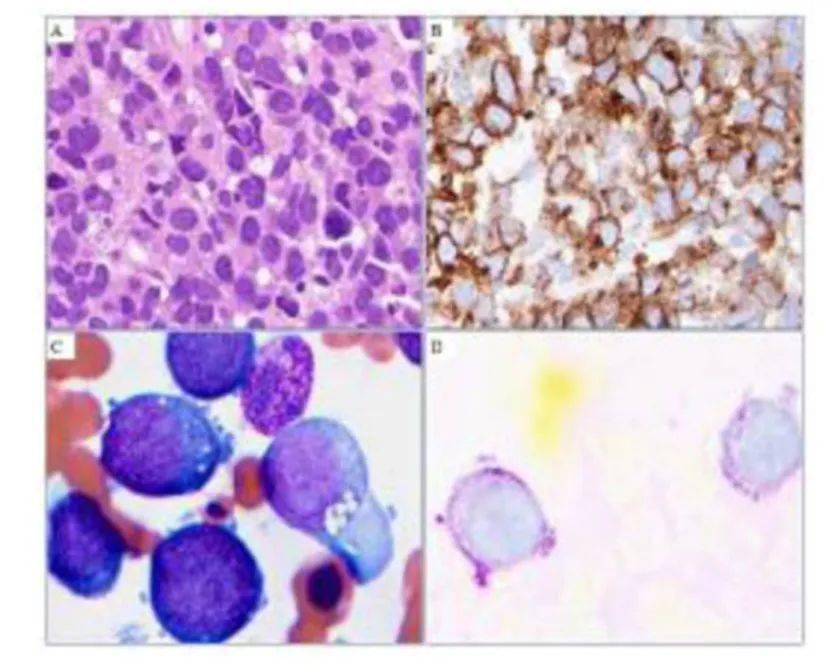
图:典型PEL骨髓象,B CD71标记, D PAS染色 来源:BLOOD 相关转录因子、分子突变和表观遗传学 GATA1和PU.1 GATA1和PU.1作为两个相互拮抗的谱系特异性转录因子,调节红系和髓系分化,其中GATA1在红系存活和终末分化中起关键作用。 体外研究表明,PU.1可抑制PEL细胞株的生长并导致凋亡,其机制与促癌基因如c-MYC和BCL2的下调以及GATA1转录因子DNA结合活性降低有关。外源性GATA1 通过与PU.1启动子及其调控上游反应元件结合来抑制PU.1的表达,从而 导致PU.1阻断的小鼠红白血病细胞继续分化。 GATA-1介导机制在人和小鼠的红白血病细胞系中表现出明显的种间差异,与小鼠AML-EL不同,人GATA1对PU.1的抑制作用还涉及DNA结合以及调节上游增强子和启动子区域的H3K9和H3K27三甲基化,提示了去甲基化药物潜在治疗靶点。 TP53 Rose等检测了166例PEL患者分子突变情况,发现TP53是PEL唯一的高频基因,对比其他AML类型为36% vs 11%。而相应其它AML常见的基因突变,如ASXL1、DNMT3A、FLT3-ITD、IDH2、NPM1、NRAS、RUNX1和TET2相应少见。Montalban-Bravo和Benton等人最近报告显示:>90%的PEL患者中发现TP53异常(包括突变和17p-),提示TP53在PEL的疾病转化中的重要作用。 铁/血红素稳态和p53信号通路之间似乎存在联系,在铁过载时p53下调,通过复杂机制影响到p53核输出、p53稳态和DNA相互作用,铁诱导的DNA损伤,通过氧自由基介导,最终导致了TP53高频突变和复杂核型形成。 GATA1和p53相互作用 GATA1直接影响p53途径:作用于p53转录活化元件并抑制其在红系前体细胞中转录功能,并具有红系特异性。GATA1是红细胞分化关键分子,GATA1基因敲除可诱导小鼠PEL,红系持续恶性增殖累及导致二级突变发生,同时GATA1敲除后对p53抑制作用减弱,导致了白血病克隆的扩增。 c-MYC和 溴结构域蛋白(BRD) BRD家族是组蛋白修饰蛋白,具有“读取”基因的能力,并通过招募转录调节因子到特定基因位点来调控基因表达。BRD蛋白家族由四种同源蛋白组成:BRD2、BRD3、BRD4和BRDT,在细胞周期生长和调控中具有广泛的作用,是表观遗传靶点。BRD可以促进并维持异常基因表达,其中就涉及到MYC基因的持续表达。 GFI-1B和LSD-1 PEL另一个重要的转录因子是生长因子非依赖1B蛋白(GFI-1B),其在红系祖细胞生长和分化诱导中起着关键作用,GFI-1B的过度表达仅限于AML-M6和AML-M7亚型,并且与祖细胞系增殖能力的增加有关。siRNA沉默可降低体外红系白血病细胞的增殖能力。GFI-1B与组蛋白去甲基化酶(LSD1)相互作用,抑制GBI-1B靶基因,从而导致谱系特异性细胞的分化。 KIT受体配体系统 一个由谱系特异性转录因子组成的系统,在红系和巨核细胞分化的交叉点上起着决定性的作用。红系和巨核细胞在调控转录因子和细胞表面标记表达上具有早期谱系同源性,KIT受体表达与谱系特异性抗原的表达有关,并决定红系分化的表型。 其它参与红系分化的转录因子: RUNX1和KLF1, GATA2,SCL/TAL,NF-E2,核因子B,FOXO,EKLF。 虽然这些因子在动物模型中发现肿瘤转化机制,但是编码这些转录因子的基因突变并未发现直接与人PEL相关。
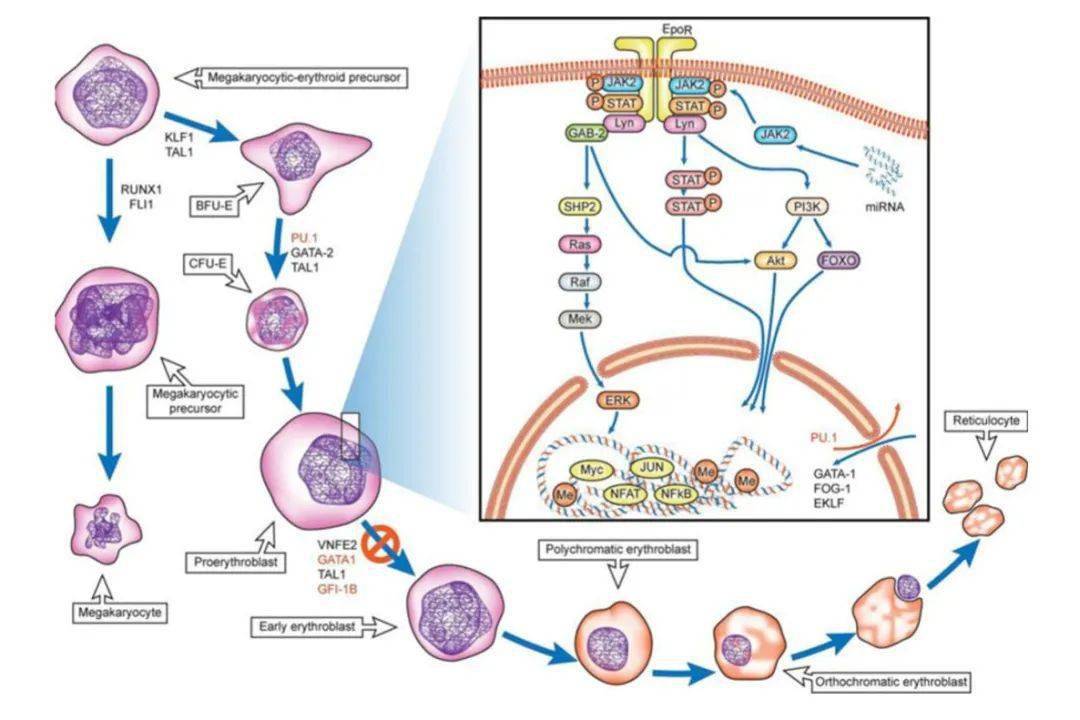
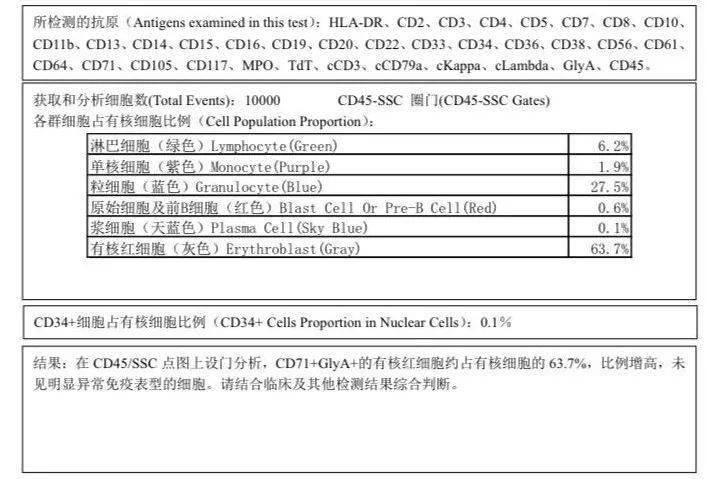
图示:人/小鼠PEL分化模型及途径 PEL的预后和治疗 PEL中位生存期为1-3个月,临床独立预后风险包括细胞遗传学、低白蛋白血症和LDH升高。 由于PEL与MDS的相关性,目前标准诱导方案是选择传统AML联合化疗还是去甲基化治疗一直存在争论,大部分研究表明两者之间没有生存差异,近期针对217例PEL临床结果回顾性研究提示,联合化疗在ORR优于去甲基化药物,但是不影响FPS和OS,但是针对高危遗传学异常的患者,显示去甲基化药物对改善OS有利。虽然大部分的患者无法进入移植阶段,但是基于目前的证据,异基因造血干细胞移植可以改善PEL的预后,同时,靶向TP53以及相关分子通路的靶向抑制剂是一种潜在的治疗选择。 Iacobucci等人利用二代测序技术对PEL的基因组结构进行了更详细的探索,涉及基因包括:细胞周期/肿瘤抑制,粘蛋白复合物形成,RNA剪接、转录、信号传导等多个基因突变,DNA甲基化和染色质修饰。此外,近半数病例中发现存在嵌合型融合。进一步体外模拟,发现仅NUP98-JARID1A融合蛋白的表达导致白血病发生,提示需要其它基因损伤的共存。此外,儿童和成人PEL的突变模式也不同,NUP98、PTPN11、GATA1和UBTF突变在儿童更为频繁,TP53和MLL突变在成人中占主导地位。33%的患者存在信号通路基因突变,体内和体外研究发现了潜在的靶向治疗位点:ALK突变的靶向药物crizotinib, NTRK1的酪氨酸激酶突变的靶向药物 entrectinib和 JAK-STAT、mTOR、PI3K途径的靶向药物JAK2抑制剂 ruxolitinib。 浙大二院血液科典型病例分享 CASE 1 2018年起病 患者,男性,57岁 主诉:乏力头晕1月 辅助检查 血常规:白细胞3.7×10^9/L↓、血红蛋白56g/L↓、血小板计数45×10^9/L↓、幼稚细胞1.0%,肝肾功能基本正常,乳酸脱氢酶593U/L↑、凝血功能正常,ANA阴性,铁蛋白升高520.2μg/L↑。 初次骨髓检查 提示增生性贫血,可见有病态造血,涂片原始细胞比例2%,免疫分型提示有核红细胞63.7%,但未见异常表型细胞,常见31种白血病融合基因检测阴性,TP53基因突变阳性。PNH及溶血性贫血检测均为阴性。
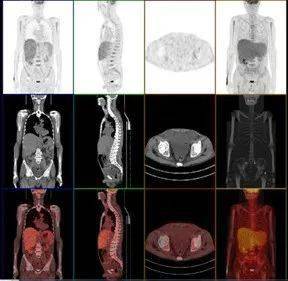
初步诊断骨髓增生异常综合征?对症支持治疗效果不佳,同时患者出现感染伴不可控性贫血和血小板减少。 PET-CT检查 全身骨骨髓糖代谢弥漫性增高、肝脾肿大伴代谢增高。
2周后再次评估骨髓
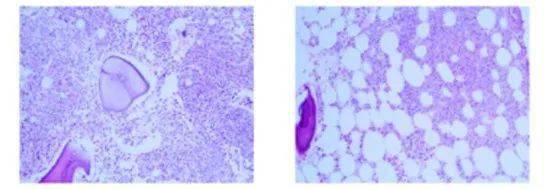
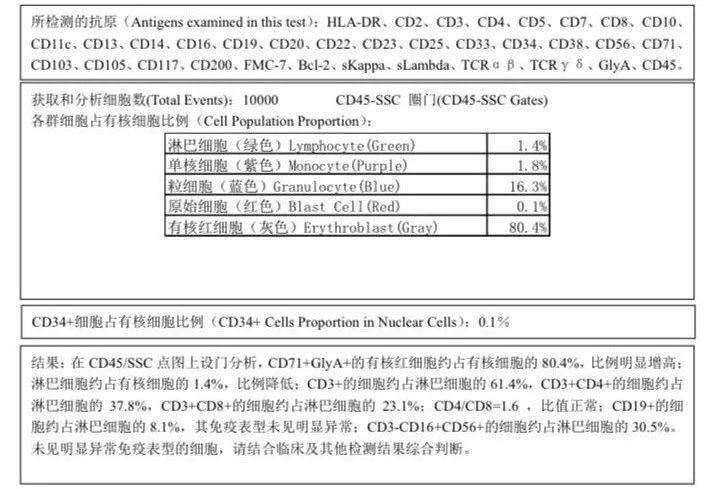
骨髓病理 髓腔内可见局灶性大而异形肿瘤细胞浸润,免疫组化提示幼稚细胞为原始红细胞,CD42b 巨核细胞+,CD235a 弥漫+,Ki-67 90%,MPO 散在+,CD99 +。 再次骨髓免疫分型:有核红细胞占有核细胞80.4%
染色体检查:复杂核型
最后诊断:纯红白血病 治疗经过 病情进行性恶化, 白细胞计数6.3×10^9/L、血红蛋白66g/L↓、血小板计数20×10^9/L↓、网织红细胞计数(%)12.57%↑、晚幼红细胞110.00个/100WBC。 感染控制不佳,降钙素原(PCT)30.698ng/mL↑,同时出现肝肾功能不全: 肌酐181μmol/L↑、尿素氮44.30mmol/L↑、总胆红素168.7μmol/L↑、直接胆红素119.4μmol/L↑、间接胆红素49.3μmol/L↑、白蛋白26.6g/L↓、谷丙转氨酶73U/L↑、谷草转氨酶149U/L↑。 反复发热,予以联合抗感染治疗效果不佳,胸闷气急明显,血氧饱和度不能维持,有气管插管指征,患方放弃插管,要求自动出院。 CASE 2 2013年起病 患者,男性,29岁 主诉: 乏力1月余,发现全血细胞减少10天。 辅助检查 血常规:白细胞计数(WBC)1.8×10^9/L↓、血红蛋白(HGB)53g/L↓、血小板计数(PLT)93×10^9/L↓ 骨髓检查 骨髓常规提示红系占57%,红系、巨核系病态造血明显,原始细胞29.8%(NEC)。 免疫分型 有核红细胞65%,原始细胞7%(NEC20%),表达HLA-DR,CD13,CD15,CD33,CD34,CD38,CD56,CD117,CD123 31项融合基因阴性,4项AML常见预后突变阴性,染色体正常核型。 WT1 1.64*10^3拷贝数。 诊断:急性红白血病(AML-M6) 治疗经过 HAG方案化疗1周期,治疗后复查骨髓:原始细胞1%,有核红细胞70%,残留病灶0.35%(有核红细胞60%),WT1 6.01*10^3copy。随后 HA方案化疗1周期(Hom 4mg d1-5,Ara-C 200mg d1-7),复查骨髓:粒系成熟欠佳与红系轻度病态生成,原始细胞0.5%,残留病灶1.98%,WT1阳性(1.57*10^3)。 2次化疗未缓解,有异基因造血干细胞移植指征。予BuCy方案预处理:Busulphan 3.2mg/kg, d-7~d-4; CTX 60mg/kg, d-3~d-2。回输非亲缘HLA全相合异基因外周造血干细胞(供者血型O型Rh+,受者A型Rh+),共回输干细胞量200ml,单个核细胞计数7.7*10^8/kg,CD34+细胞计数5.4*10^6/kg;移植后予环孢素+骁悉+短疗程MTX预防GVHD,d+11粒系植活及d+15血小板植活。定期复查骨髓常规、残留病灶阴性。 随访至2020年9月(生存7年)存活。 文献来源: Erythroleukemia-Historical perspectives and recent advances in diagnosis and management.(MDACC)BLOOD返回搜狐,查看更多 |

【本文地址】